


Fluorescent microscopy is not a transparent window but a powerful translation; its true “art and science” is the disciplined practice of managing inherent signal ambiguity. The glow you see represents both biological reality and technical artifact—distinguishing between them requires a systematic understanding of the trade-offs.
Visualizing the Fundamental Trade-Off: The Photon Budget
Every fluorescent experiment operates under a fundamental constraint: fluorophores emit a finite number of photons before bleaching. How you manage this “photon budget” determines whether you collect meaningful data or artifacts.
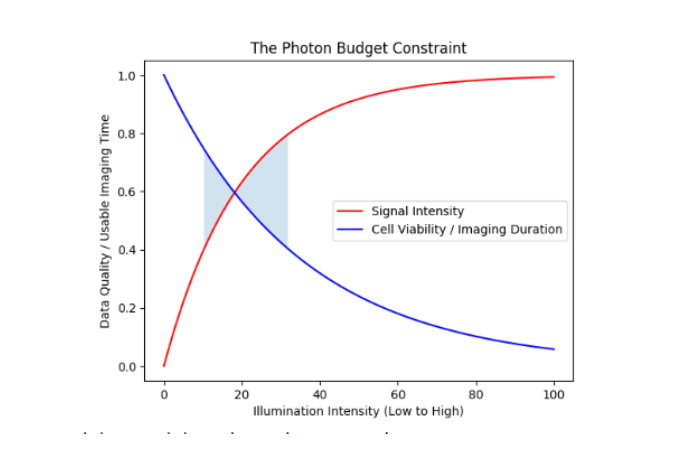
The Complete Framework: Science, Art, and Practical Implementation
The following tables systematically break down each component of fluorescent microscopy, moving from core science to practical application.
The Scientific Components & Their Hidden Ambiguities
| Component | Primary Function | Key Parameter | Hidden Ambiguity/Caveat | Critical Question to Ask |
| Fluorophore | Light-emitting molecule | Extinction coefficient, quantum yield | Finite photon budget; photobleaching is irreversible | “Am I maximizing signal through preparation rather than just increasing laser power?” |
| Excitation Source | Provide excitation light | Intensity, wavelength stability | Phototoxicity: light damages living samples | “Is my observation altering the biology I’m trying to study?” |
| Optics & Objective | Collect and focus light | Numerical Aperture (NA), magnification | Shallow depth of field creates 2D illusion from 3D reality | “What’s happening above and below my focal plane that I’m missing?” |
| Filters & Dichroics | Separate excitation from emission | Bandwidth, transmission efficiency | Spectral bleed-through (crosstalk) between channels | “Is my ‘specific’ signal contaminated from other fluorophores?” |
| Detector (Camera/PMT) | Convert photons to signal | Gain, read noise, quantum efficiency | Gain amplifies noise alongside signal | “Is this dim structure real signal or amplified noise?” |
| Sample Environment | Biological context | Refractive index, temperature, pH | Autofluorescence creates false positives | “Have I imaged an unlabeled control under identical conditions?” |
References & Further Reading: Fluorescent molecules soak up light energy, get excited, and spit it back out at a different wavelength—think absorbing blue and beaming green. The microscope’s the maestro, wielding filters and beams to catch that shift.
The Interpretive “Art” – From Design to Analysis
| Process Stage | Common Misconception | Reality (The True “Art”) | Practical Framework | Example Failure Mode |
| Experimental Design | “Controls are just for publication” | Pre-visualization of all possible artifacts | The Control Matrix: Run autofluorescence, secondary-only, and positive controls in parallel | Mistaking tissue autofluorescence for specific labeling |
| Sample Preparation | “More label = better signal” | Optimization for specificity over intensity | Titration approach: Find the minimum label concentration that gives a detectable signal above controls | Non-specific binding creating false localization patterns |
| Image Acquisition | “Maximize all settings for best image” | Strategic trade-off management | The Triangle Constraint: Choose 2: Speed, Resolution, Sensitivity | Photobleaching the entire sample before completing the time series |
| Live-Cell Imaging | “Just use lower light” | Balancing observation with perturbation | Viability vs. Visibility Index: Establish metrics for cell health during imaging | Documenting stress responses rather than normal physiology |
| Image Processing | “Processing reveals hidden truth” | Transparent enhancement of existing data | The Process Receipt: Document every adjustment with software and parameters | Creating structures through excessive deconvolution or thresholding |
| Data Interpretation | “Colocalization = interaction” | Hypothesis generation, not conclusion | The Resolution Reality Check: 200nm separation can appear as colocalization | Claiming protein interactions without biochemical validation |
The Ambiguity Audit Framework – A Step-by-Step Checklist
| Audit Phase | Specific Questions | Documentation Required | Acceptable Outcome | Red Flag |
| 1. Pre-Acquisition Design | – What are my 3 most likely artifacts? – Have I designed controls for each? – What is my photon budget strategy? |
Written experimental protocol with control rationale | Clear mapping of potential artifacts to specific controls | No biological replicates; single control for multiple conditions |
| 2. Acquisition Parameters | – Are my settings reproducible? – Am I in the linear detection range? – Have I minimized observer bias? |
Saved instrument settings file; metadata with images | Settings allow reproduction by trained colleague | Gain set above manufacturer’s linear range recommendation |
| 3. Control Validation | – Do controls look as expected? – Is signal-to-noise > 3:1? – Is autofluorescence < 10% of signal? |
Images of all controls with same display settings | Controls show expected negative/positive results | Autofluorescence pattern mimics “specific” signal |
| 4. Processing Transparency | – Can I list every adjustment? – Were identical adjustments applied to all compared images? – Does raw data support processed conclusions? |
Processing workflow with software name, version, and parameter values | Raw data visible alongside processed version | Adjustments that “create” data not visible in raw image |
| 5. Interpretation Boundaries | – What are 2 alternative explanations? – Does my conclusion respect optical limits? – What further experiment would confirm? |
Written interpretation with acknowledged limitations | Conclusions are conservative and suggest next experiments | Claiming molecular interactions from standard resolution images |
Application-Specific Guidelines & Trade-Offs
| Application | Primary Goal | Optimal Technique | Critical Trade-Off | Common Pitfall | Validation Required |
| Protein Localization | Subcellular targeting | Widefield or confocal | Resolution vs. throughput | Misinterpreting overexpression artifacts | Biochemical fractionation + orthogonal method |
| Live-Cell Dynamics | Temporal tracking | Spinning disk confocal or light sheet | Temporal resolution vs. phototoxicity | Observing stress responses instead of physiology | Cell viability assays post-imaging |
| Super-Resolution | Nanoscale detail | STORM/PALM or STED | Resolution vs. sampling/throughput | Reconstruction artifacts from sparse sampling | Resolution measurement on biological structure |
| Multicolor Imaging | Interaction or proximity | Spectral imaging or careful filter sets | Number of channels vs. crosstalk | Spectral bleed-through misinterpreted as colocalization | Unmixing controls and spectral validation |
| Quantitative Intensity | Concentration measurements | Widefield with careful calibration | Signal linearity vs. dynamic range | Ignoring non-linear detector response | Standard curves with known concentrations |
| Clinical/Diagnostic | Reproducible detection | Standardized widefield | Sensitivity vs. specificity | Inter-operator variability | CLIA-type validation with positive/negative controls |
The Evolution of “Art” with Computational Advances
| Traditional Skill | Computational Equivalent | New Ambiguity Introduced | Required New “Art” Skill | Quality Control Metric |
| Manual focus optimization | Automated focus algorithms | Algorithm failure on heterogeneous samples | Understanding algorithm failure modes | Visual verification of focus across field |
| Visual filter selection | Spectral unmixing | Mathematical solutions not biologically valid | Recognizing physically impossible unmixing results | Validation with single-label controls |
| Expert thresholding | Machine learning segmentation | Training set bias | Critical evaluation of training data representativeness | Performance on diverse, unseen test images |
| Judicious deconvolution | AI-based restoration | Hallucination of plausible structures | Distinguishing between restoration and invention | Comparison with physical resolution limits |
| Qualitative assessment | Quantitative feature extraction | Statistical vs. biological significance | Understanding multiple testing and effect sizes | Power analysis and false discovery rates |
Synthesis: The Integrated Practitioner’s Mindset
The tables above reveal that fluorescent microscopy expertise isn’t a linear progression but a simultaneous management of multiple dimensions:
- The Resource Dimension(Photon budget, time, sample viability)
- The Artifact Dimension(Autofluorescence, bleed-through, noise)
- The Interpretation Dimension(Optical limits, statistical significance)
- The Technological Dimension(Tool capabilities and limitations)
Expert practitioners don’t avoid these constraints—they map them systematically. The “art” is knowing which constraints are flexible for a given biological question and which are absolute boundaries.
Practical Implementation: Your First-Experiment Roadmap
For beginners, follow this prioritized sequence:
1. Week 1-2: Master Controls
- Imagine your sample type without any labels (autofluorescence baseline)
- Optimize fixation/permeabilization to minimize background
- Establish what “zero” actually looks like in your system
2. Week 3-4: Single-Channel Optimization
- Titrate your primary label to find the minimum concentration for detection
- Document the exact relationship between laser power and bleaching rate
- Establish your system’s practical signal-to-noise limits
3.Week 5+: Add Complexity Gradually
- Add the second channel only after the first is fully characterized
- Validate separation empirically, not just from filter specifications
- Always maintain the control disciplines established in Week 1
Conclusion: Beyond the Binary
The “art and science” framework is misleading if seen as separate domains. The science reveals what’s technically possible; the art navigates what’s biologically meaningful within those technical constraints. Your growing expertise is reflected not in making prettier images, but in making more truthful claims with appropriately increasing complexity.
The most sophisticated practitioners are distinguished by their systematic documentation of the journey from raw photons to biological conclusion—a chain of evidence where each link’s strength and limitations are explicitly acknowledged. This transparency, more than any image’s beauty, represents the true integration of art and science in illuminating the invisible.
FAQs
Q1: What’s the single most important control experiment in fluorescent microscopy?
A: The autofluorescence control—imaging your sample without any fluorescent labels under identical conditions. This establishes the baseline glow of your system, without which you cannot claim any specific signal.
Q2: Can I trust colocalization (yellow in merged images) to indicate protein interactions?
A: No. Due to diffraction limits (~250nm), proteins up to 200nm apart appear colocalized. Yellow indicates proximity within optical limits, not molecular interaction, which requires validation with techniques like FRET.
Q3: How do I choose between widefield and confocal microscopy?
A: Widefield is better for dim samples, rapid dynamics, and quantitative intensity measurements. Confocal is superior for thick samples, reducing out-of-focus light, and optical sectioning. The choice involves trading light collection efficiency for optical sectioning capability.
Q4: Why do my live cells die during imaging, and how can I prevent it?
A: Cell death typically comes from phototoxicity (light damage). Prevention strategies include: using lowest possible light intensity, imaging less frequently, using specialized techniques like spinning disk confocal, and employing oxygen scavengers to reduce photodamage.
Q5: Are AI-enhanced images from computational microscopy “real”?
A: They are mathematical reconstructions based on probability. While they can reveal structures below diffraction limits, they can also generate plausible artifacts. Validation against physical benchmarks and biological context remains essential—the algorithm’s output requires the same level of skepticism as traditional images.